Radiobiologia komórkowa
Zbadano zależności między sygnalizacją rozpoczynaną przez EGFR (receptor epidermalnego czynnika wzrostowego), łączeniem dwuniciowych pęknięć DNA (DSB) i przeżywalnością po ekspozycji na promieniowanie X komórek ludzkiego glejaka M059 linii K i J. Linie te różnią się promieniowrażliwościa (ryc. 1): linia M059K jest stosunkowo promieniooporna, linia J jest promieniowrażliwa, z defektem łączenia DSB z powodu braku podjednostki katalitycznej kinazy białkowej zależnej od DNA, DNA-PKcs. Komórki traktowano inhibitorem sygnalizacji, tyrfostyną AG 1478, swoistą dla aktywności kinazowej EGFR. Następnie komórki napromieniano promieniowaniem X. Przeżywalność mierzono testem klonogenności, łączenie DSB monitorowano elektroforezą pulsową (PFGE).
Jak widać na ryc. 2, w napromienionych komórkach M059 K aktywność kinazowa EGFR była potrzebna do skutecznego łączenia DSB (tyrfostyna AG 1478 działała promieniouczulająco w zakresie dawek obniżających przeżywalność do 70-80 %), natomiast zahamowanie kinazy EGFR nie powodowało promieniouczulenia i nie wpływało na łączenie DSB w komórkach M059 J z defektem DNA-PKcs. Ponadto translokację EGFR do jądra komórkowego (mierzoną metodą ELISA) obserwowano po napromienieniu w komórkach M059 K, lecz nie M059 J (ryc. 3). Dane te sugerują wpływ aktywności kinazowej EGFR na zależne od DNA-PK łączenie DSB a z drugiej strony - rolę DNA-PKcs w translokacji EGFR do jądra komórkowego, prawdopodobnie przez ko-translokację z podjednostkami DNA-PKcs z puli cytoplazmatycznej.
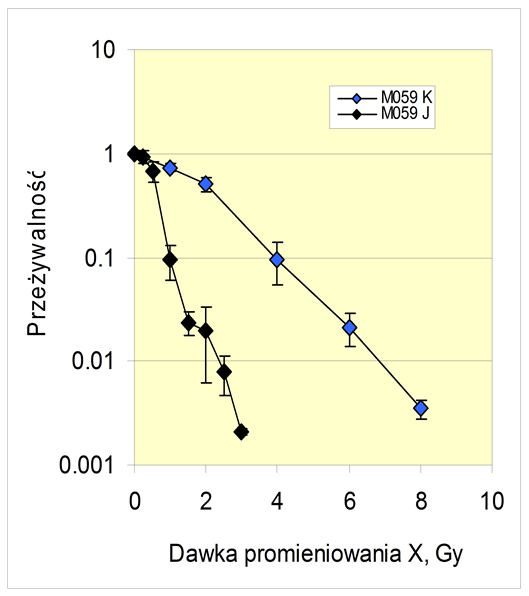
Ryc. 1. Krzywe przeżywalności 2 linii komórek glejaka M059.
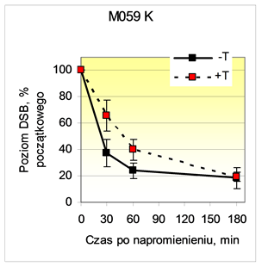
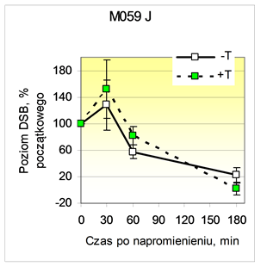
Ryc. 2. Łączenie indukowanych przez promieniowanie X pęknięć dwuniciowych DNA (DSB) w komórkach M059 K i M059 J nie traktowanych (-T) lub traktowanych wstępnie (+T) tyrfostyną AG 1478, inhibitorem aktywności kinazy tyrozynowej EGFR.
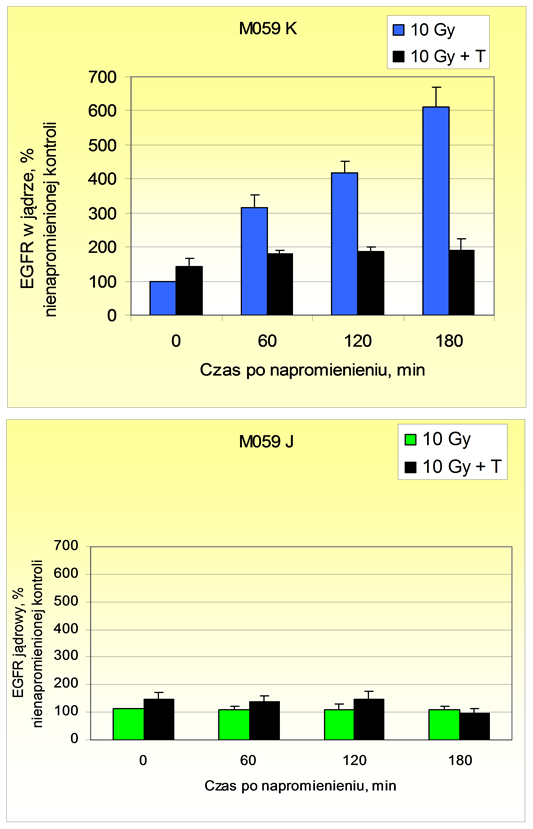
Ryc. 3. Nagromadzenie EGFR w jądrach komórkowych komórek M059 K I J po ekspozycji na promieniowanie X (10 Gy), w obecności lub nieobecności tyrfostyny AG 1478 (T). Stosowano równotoksyczne stężenia inhibitora, tzn. 4,5 μM (M059 K) i 9 μM (M059 J). Średnie wartości + odchylenia standardowe z 2 doświadczeń).